Поиск по сайту
Наш блог
Это странная ситуация: вы соблюдали все меры предосторожности COVID-19 (вы почти все время дома), но, тем не менее, вы каким-то образом простудились. Вы можете задаться...
Как диетолог, я вижу, что многие причудливые диеты приходят в нашу жизнь и быстро исчезают из нее. Многие из них это скорее наказание, чем способ питаться правильно и влиять на...
Овес-это натуральное цельное зерно, богатое своего рода растворимой клетчаткой, которая может помочь вывести “плохой” низкий уровень холестерина ЛПНП из вашего организма....
Если вы принимаете витаминные и минеральные добавки в надежде укрепить свое здоровье, вы можете задаться вопросом: “Есть ли лучшее время дня для приема витаминов?”
Ты хочешь жить долго и счастливо. Возможно, ты мечтал об этом с детства. Хотя никакие реальные отношения не могут сравниться со сказочными фильмами, многие люди наслаждаются...
Приседания и выпады-типичные упражнения для укрепления мышц нижней части тела. Хотя они чрезвычайно распространены, они не могут быть безопасным вариантом для всех. Некоторые...

Ученые из Стэнфордского университета разработали программу предсказывающую смерть человека с высокой точностью.

Глава Минздрава РФ Вероника Скворцова опровергла сообщение о падении доходов медицинских работников в ближайшие годы. Она заявила об этом на встрече с журналистами ведущих...

Федеральная служба по надзору в сфере здравоохранения озвучила тревожную статистику. Она касаются увеличения риска острой кардиотоксичности и роста сопутствующих осложнений от...

Соответствующий законопроект внесен в палату на рассмотрение. Суть его заключается в нахождении одного из родителей в больничной палате бесплатно, в течении всего срока лечения...

- Категория: Секреты неврологии
1. Что происходит в пресинаптическом окончании двигательного нервного волокна во время нервно-мышечной передачи?
Когда волна деполяризации (потенциал действия) проходит по двигательному волокну и достигает его окончания (ирссипиитической терминалы), открываются потенциал-зависимые кальциевые каналы, делая возможным входящий ток ионов кальция (Са2+). Это запускает процесс слияния везикул, содержащих ацетилхолин (АХ) с мембраной и высвобождение ацетил холи на в пространство между невраль-ной и мышечной мембранами (сипиитическую щель) (рис.).
2. Что происходит в мышце (ностсинаптичсских структурах) во время нервно-мышечной передачи?
Связывание двух молекул ацетилхолина с каждым ацетилхолиновым рецептором мышечной (постсинаптической) мембраны приводит к открытию Na* канала внутри рецептора, делая возможным входящий ток ионов натрия (Na*), который генерирует субпороговую деполяризацию, называемую миниатюрными потенциалами концевой пластинки (МПКП). МПКП каждого мышечного волокна суммируются, формируя потенциал концевой пластинки (ПКП) данного волокна. Когда одновременно активируется критическое количество рецепторов, потенциал концевой пластинки становится достаточно большим для того, чтобы вызвать потенциал действия, который затем распространяется вдоль мышечной саркоплазматической мембраны к системе Т-трубочек, приводя к высвобождению Са2* из саркоплазмати-ческого ретикулума и запуская мышечное сокращение.
3. Что происходит в синантической щели во время нервно-мышечной передачи?
После того как молекулы АХ связались с рецептором и активировали его, они освобождаются обратно в синаттическую щель, где ацетилхол инэстераза в течение долей миллисекунды расщепляет ацетилхолин на холин и уксусную кислоту. Холин подвергается обратному захвату лресинаптическим нервным окончанием и используется для синтеза нового ацетилхолина при помощи фермента холннацетилтрансферазы.
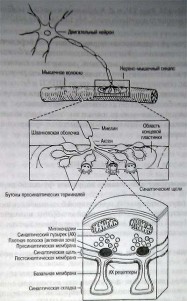
Нервно-мышечный синапс
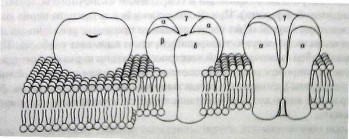
Молекулярная структура ацетилхолинового рецептора
5. Что такое -«резерв надежности» нервно-мышечной передачи?
У здорового человека количество ацетилхолина, высвобождаемого из ггреси-наптического нервного окончания, медленно уменьшается с каждой повторной деполяризацией нерва. Это значит, что активируется меньше рецепторов в концевой пластинке мышцы, генерируется меньшее число МПКП и более низкий ПКП, Однако количество рецепторов все еще достаточно велико для того, чтобы ото небольшое снижение высвобождения ацетилхолина не препятствовало достижению ПКП порога деполяризации мышечного волокна и полноценному мышечному сокращению. Эта функциональная избыточность и называется «резервом надежности» нервно-мышечной передачи.
![]()
- Категория: Секреты неврологии
58. Что такое злокачественный нейролептический синдром (ЗНС)?
3НС—угрожающее жизни осложнение лечения нейролептическими средствами, такими как фенотиазины, галоперидол, клозапин. Летальность при нем достигает 30%. Хотя чаще всего ЗНС встречается на ранних этапах лечения, симптомы могут возникать в любое время на протяжении лечения. ЗНС характеризуется лихорадкой, мышечной ригидностью, увеличением активности КФК и рабдомиолизом. Возможно развитие делирия и вегетативных нарушений. ЗНС может начинаться только с одного из вышеперечисленных симптомов. Важно распознать это состояние как можно раньше, поскольку рано начатое лечение может спасти жизнь пациенту.
59. Каковы причины ЗНС? Как он лечится?
Причина ЗНС недостаточно ясна. Предполагают, что он развивается вследствие блокады дофаминовых рецепторов в гипоталамусе. Некоторые эксперты считают, что ЗСН возникает в результате непосредственного влияния на вхождение кальция в мышечные волокна, что приводит к интенсивным мышечным сокращениям и гиперметаболическому состоянию, сходному с таковым при злокачественной гипертермии. Лечение включает отмену препарата, вызвавшего ЗСН, назначение бромокриптина, 5 мг 3 раза в день, или дантролена, 0,5-3 мг внутривенно один раз в день. Целесообразно наблюдение в условиях отделения интенсивной терапии.
60. Какие миопатии связаны с птозом или офтальмоплегией?
Птоз, обычно не сопровождающийся офтальмоплегией:
- миотоническая дистрофия
- врожденные миопатии
- центронуклеарная миопатия
- немалиновая миопатия
- миопатия центрального стержня
- десминовая миопатия.
Птоз, сопровождающийся офтальмоплегией:
- окулофарингеальная мышечная дистрофия
- окулофарингодистальная миопатия
- хроническая прогрессирующая наружная офтальмоплегия (митохондри-альная миопатия).
61. Какие миопатии характеризуются слабостью преимущественно дистальных мышц?
- дистальная миопатия 1-го типа с началом в зрелом возрасте (Веландер)
- дистальная миопатия 2-го типа с началом в зрелом возрасте (Марксбери) или тибиальная мышечная дистрофия (Удда)
- дистальная миопатия 1-го типа с началом в молодом возрасте (Нонака)
- дистальная миопатия 2-го типа с началом в молодом возрасте (Миоши)
- дистальная миопатия 3-го типа с началом в молодом возрасте (Лейнга)
- дистальная дистрофинопатия с началом в зрелом возрасте
- дистальная миопатия с началом в детском возрасте
- миотоническая дистрофия
- лицелопаточно-плсчевая дистрофия
- скапулопероиеальиая миопатия
- окулофариигеальная дистрофия
- мышечная диагрофия Эмери-Дрейфуса
- воспалительные миопатий; миозит с включениями
- метаболические миопатии: недостаточность фермента, уменьшающего ветвление гликогена? недостаточность кислой мальтазы
- врожденные миопатий: иемалииовая миопатия, миопатия центрального стержня, центронуклеарная миопатия.
- десминовая (миофибриллярлад) миопатия
62. Каковы характерные признаки миофибриллярных миопатий?
- Клинические признаки: медленно прогрессирующая слабость, обычно вовлекающая как дистальиые, так и проксимальные отделы, изредка—парестезии, атрофия мышц, напряжение и болезненные спазмы мышц, миалгии. В редких случаях первым проявлением бывает дыхательная недостаточность. У 50% пациентов выявляется кардиомиопатия. ЭМГ обычно выявляет изменения, характерные для миопатий и/или денервации. Уровень сывороточной КФК может быть нормальным или превышает норму не более чем в 7 раз.
- Патоморфологические признаки: деградация (фокальная), преимущественно затрагивающая миофибриллы, лизис актина, а-актинина ± титина, небулина или миозина, неадекватная экспрессия киназы цикла деления клетки 2 и циклин-зависимых киназ 2,4, 8 и 7. Морфологическим маркером являются гиалиновые сфероидные структуры, которые содержат компактно упакованные разрушенные мисфибриллярные структуры и интенсивно воспринимают окраску на актин. В местах повреждений и мышечных волокнах накапливаются многочисленные белки: десмин, ламин В, гелсолин, убиквитин, al-антихимотрипсиновая молекула адгезии нервных клеток, N-концевой бета-амилоид, эктопический дистрофии, у-саркогликан и амилоидные отложения. Обнаружены мутации в генах, кодирующих десмин и альфаВ-кристаллин.
- Категория: Секреты неврологии
53. Что такое -«синдром ригидного человека»?
Синдром ригидного человека характеризуется флуктуирующим напряжением аксиальных мышц и проксимальных мышц конечностей, на которое «накладываются» внезапные мышечные спазмы. Симптомы усиливаются эмоциональными, соматосенсорными или звуковыми стимулами. У многих пациентов выявляются аутоиммунные эндокринопатии, наиболее часто - инсулинозависимый сахарный диабет. В сыворотке и ЦСЖ выявляют антитела против глутаматдекарбоксилазы -фермента, участвующего в синтезе ГАМК. ЭМГ выявляет постоянные низкочастотные разряды нормальных потенциалов двигательных единиц, которые регистрируются и в покое. В некоторых случаях, связанных со злокачественными новообразованиями, антитела к глутаматдскарбоксилазе отсутствуют. Вместо этого у них выявляются антитела против антигена 128-кд, известного как амфифизин. Значительное симптоматическое улучшение достигается при приеме внутрь бензо-диазепинов, прежде всего диааепама (10-100 ДО/день). Для ослабления симптомов возможно также применение баклофена и валъпроевой кислоты. У части больных улучшения можно добиться с помощью кортикостероидов и плазмафереза. Недавние исследования продемонстрировали эффективность леветирацетама в отношении пароксизмальных симптомов.
54. Какие лекарственные средства могут вызвать воспалительную миопатию?
Болезненная воспалительная миопатия развивается у части пациентов под действием D-пенициламина или прокаинамида. Кроме того, воспалительную миопатию могут вызывать фенилбутазон, нифлюмовая кислота, пропилтиоурацил, пенициллин, сульфаниламиды, циметидин, симвастатин, кокаин.
55. Перечислите наиболее частые миотоксические препараты.
- Клофибрайт и другие гиполипидемические средства
- Хлорокин
- Эметин
- Алкоголь
- Эпсилон-аминокапроновая кислота
- D-пенициламин
- Фенформин
- Зидовудин
56. Пациент со СПИДом, который принимает зидовудин, жалуется на миалгию и слабость. Назовите возможные причины.
Точная диагностика в данном случае затруднена. Миалгия и увеличение активности КФК часто наблюдаются при СПИДе, причем у некоторых больных развивается симметричная преимущественно проксимальная мышечная слабость. При ЭМГ выявляются те же признаки, что и при полимиозите. При мышечной биопсии могут обнаруживаться изменения, типичные для полимиозита (некротические волокна с перимизиальной, эндомизиальной и периваскулярной лимфоцитарной инфильтрацией). Зидовудин также способен вызвать миопатию, которая характеризуется главным образом атрофией мышц и проксимальной слабостью. Обычно миопатия развивается у пациентов, которые принимали высокие дозы препарата свыше б месяцев. При исследовании биоптата мышцы в этом случае выявляются изменения, характерные для патологии митохондрий, например могут отмечаться многочисленные -«рваные красные волокна». Встречаются также палочки (немалиновые) и цитоплазматические включения. Клинические признаки миопатии, а также морфологические изменения, выявляемые при биопсии, могут уменьшаться после прекращения приема зидовудина. Предполагают, что миопатия связана со способностью препарата ингибировать митохондриальную ДНК-полимеразу и таким образом нарушать функционирование митохондриальной ДНК. Тем не менее другие антиретровируоные вещества, используемые для лечения ВИЧ-инфекции, такие как диданозин и аальцитабин, являясь более сильными ингибиторами митохондриальной функции, не вызывают подобные симптомы. Таким образом, по-видимому, развитие миопатии объясняется другими, пока неизвестными факторами.
57. Что такое стероидная миопатия?
Существуют две формы стероидной миопатии. Наиболее распространенная форма характеризуется прогрессирующей мышечной слабостью без болевого синдрома. Обычно миопатия связана с длительным применением кортикостероидов, однако при применении ингаляционных препаратов слабость диафрагмы может развиваться в течение двух недель после начала лечения. Миотоксичность кортикостероидов при их длительном применении можно частично предотвратить физической нагрузкой. При снижении дозы или отмене препарата симптомы регрессируют. Уровень КФК не повышается, а ЭМГ либо не выявляет патологии, либо показывает минимальные миопатические изменения.
Вторая форма стероидной миопатии связана с приемом высоких доз кортикостероидов, обычно в сочетании с воздействием деполяризующих миорелаксантов, сепсиса и недостаточного питания. Она характеризуется остро развивающейся тяжелой мышечной слабостью, которая может захватывать все мышцы, включая дыхательные. Этому синдрому было дано много названий, в том числе острая квад-риплегическая миопатия, миопатия толстых филаментов, миопатия критических состояний. ЭМГ в дополнение к миопатическим изменениям часто выявляет признаки острой аксональной полиневропатии, что затрудняет диагностику. Симметричная проксимальная слабость и атрофия мышц развиваются в течение нескольких дней. При наличии адекватной поддерживающей терапии возможно восстановление в течение нескольких месяцев. Причиной миопатии является значительная потеря толстых миофиламентов с сохранением тонких (актиновых) филаментов и Z-дисков в атрофичных мышечных волокнах. У 30-50% пациентов наблюдается повышение уровня КФК. При наличии поддерживающей терапии прогноз восстановления вариабелен (от 1 месяца до 1 года), однако характерна значительная летальность.
![]()
- Категория: Секреты неврологии
49. Что такое миокимия?
Миокимия — постоянные волнообразные сокращения группы мышечных волокон, вызванные последовательной спонтанной активацией отдельных двигательных единиц. На ЭМГ они выявляются как группы из 10-20 потенциалов, имеющих частоту 5-60 Гц и регулярно повторяющихся с интервалом в 0,2-1 сек. Миокимия особенно часто наблюдается в мимических мышцах. Причиной миокимии могут быть поражения ствола головного мозга (особенно при рассеяном склерозе), лучевая невропатия, синдром Гийена-Барре, хронические заболевания периферических нервов, укус лесной гремучей змеи, лечение препаратами золота, синдроме Исаакса.
50. Для каких состояний характерно появление миотонических разрядов?
Миотонические разряды встречаются при врожденной миотонии, врожденной парамиотонии, миотонической дистрофии, синдроме Шварца-Джампеля, инфантильной и взрослой форме недостаточности кислой мальтазы, гиперкалиемическом периодическом параличе. Врожденная миотония возникает вследствие нарушения функции хлорных каналов, тогда как врожденная парамиотония, гиперкалиемический периодический паралич и синдром Шварца-жампеля вызваны нарушением функции натриевых каналов. Миотония при миотонической дистрофии и недостаточности кислой мальтазы вызвана другими, недостаточно ясными мембранными дефектами.
51. Что такое нейромиотония?
Нейромиотонпя — состояние, характеризующееся постоянными мышечными сокращениями и мышечным напряжением, которые объясняются повторяющимися разрядами в периферическом нерпе, возникающие отдельными вспышками. По своей природе это нейрогениый синдром, связанный с развитием в силу неясных причин гипервозбудимости двигательных нервных волокон. Причиной нейромиотонии может быть аутоиммунная нейрогеиная гипервозбудимость. Во многих случаях при нейром и отопи и выявляют антитела к потенциал-зависимым калиевым каналам. Миотония, в отличие от нейромиотонии, является миогенным расстройством. Именно поэтому кураре не способно затормозить миотонию, но может заблокировать нейромиотонию.
52. Что такое синдром Исаакса?
Синдром Исаакса был описан под несколькими названиями, в том числе мио-кимия с нарушением расслабления мышц, нейромиотония, псевдомиотония, синдром броненосца, синдром постоянной активности мышечных волокон. Жалобы включают с трудом преодолимое напряжение мышц, перемежающиеся спазмы мышц, затруднения при жевании, разговоре и даже дыхании. Наиболее заметным признаком синдрома Исаакса является миокимия.
У некоторых пациентов в сыворотке выявлены антитела, направленные против потенциал-зависимых каналов пресинаптических окончаний, которые и приводят к гипервозбудимости дистального участка двигательных волокон или нервных окончаний. ЭМГ выявляет спонтанные длительно продолжающиеся нерегулярные вспышки разрядов различной формы, которые возникают в проксимальных участках нервов. В качестве серологического маркера этого синдрома описаны антитела к потенциал-зависимым каналам пресинаптической терминали. У этих пациентов обнаружены также антитела к ацетилхолиновым рецепторам нервных ганглиев.
С симптоматической целью применяется фенитоин (300-400 мг/день) или карбамазепин (200 мг 3 или 4 раза в день). В некоторые случаях эффективны плазмаферез или внутривенный иммуноглобулин.
![]()
- Категория: Секреты неврологии
44. Как лечить периодический паралич?
Ацетазоламид, ингибитор карбоангидразы, эффективен у части пациентов с различными формами периодического паралича. Он особенно эффективно предупреждает приступы гипокалиемического периодического паралича, которые провоцируются различными факторами, снижающими уровень калия в плазме. Другой ингибитор карбоангидразы—дихлорфенамид, возможно, более эффективен, чем ацетазоламид, в предотвращении приступов и уменьшении межприступной слабости. Пациентам, которые не переносят ингибиторы карбоангидразы, может быть полезен прием калий-сберегающих диуретиков, таких как спиронолактон и триамтерен. При гипокалиемическом периодическом параличе обычно рекомендуется диета с низким содержанием углеводов и натрия. Ингаляция агониста бета-адренорецепторов сальбутамола может предотвратить приступ у части пациентов с гиперкалиемическим периодическим параличом. Употребление пищи, богатой углеводами, но с низким содержанием калия также может облегчить приступ.
45. При каких состояниях развивается гипертрофия мышц?
- некоторые мышечные дистрофии (Дюшенна, Беккера, конечностно-поясная)
- врожденная миотония
- спинальная амиотрофия
- цистицеркоз
- амилоидоз
- акромегалия
- синдром Флайера (инсулинорезистентный сахарный диабет в сочетании с черным акантозом)
- наследственные моторно-сенсорные невропатии
- хроническая (рецидивирующая) воспалительная полиневропатия
- детский тип недостаточности кислой мальтазы
- миопатия при врожденном гипотиреоидизме (синдром Кохера-Дебре-Семелена)
- гиперкалиемический периодический паралич
- фокальные мононевропатии (фокальная гипертрофия)
- радикулопатии (фокальная гипертрофия)
- саркоидоз (преимущественно фокальная гипертрофия)
46. Какие миопатии вызывают дыхательную недостаточность?
- некоторые мышечные дистрофии (Дюшенна, Беккера, конечностно-поясная, Эмери-Дрейфуса; миотоническая, врожденные)
- миопатия вследствие недостаточности кислой мальтазы
- миопатия вследствие недостаточности карнитина
- немалиновая миопатия
- митохондриальные миопатий
- центронуклеарная миопатия
- полимиозит
Учтите! Дыхательная недостаточность может быть первым проявлением заболевания.
47. Какие миопатии связаны с дисфагией?
- окулофарингеальная мышечная дистрофия
- миозит с включениями
- миотоническая дистрофия
- митохондриальные миопатии
- полимиозит и дерматомиозит
- мышечная дистрофия Дюшенна
48. Какие миопатии связаны с патологией сердца?
- Нарушения сердечного ритма—болезнь Кирнса-Сейра; синдром Андерсена; полимиозит; мышечные дистрофии: миотоническая, конечностно-пояс-ная (IB, 2С, 2D, 2Е, 2F, 2G, 21 типов), Эмери-Дрейфуса.
- Сердечная недостаточность — мышечные дистрофии: Дюшенна, Беккера, Эмери-Дрейфуса, миотоническая, конечностно-поясная (IB, 2С, 2D, 2Е, 2F, 2G, 21 типов); немалиновая миопатия; недостаточность кислой мальтазы; недостаточность карнитина; полимиозит.
КЛЮЧЕВЫЕ ФАКТЫ: МИОПАТИИ
- Миопатии проявляются симметричной слабостью проксимальных мышц, которая может сопровождаться иными симптомами
- Диагноз миопатии могут подтвердить исследование активности КФК, данные ЭМГ и мышечной биопсии
- Мышцы содержат как медленные волокна (волокна 1 го типа, или красные волокна), так и быстрые волокна (волокна 2-го типа, или белые волокна)
- Миотоническая дистрофия—самая распространенная форма мышечной дистрофии у взрослых
- Дыхательная недостаточность—наиболее серьезное возможное осложнение при лечении большинства пациентов с миопатиями
- При выяснении причины миопатии следует всегда исключать токсическое действие лекарственных средств
- Злокачественный нейролептический синдром—неотложное состояние, характеризующееся высокой летальностью
![]()
- Категория: Секреты неврологии
37. Что такое болезнь Мак-Ардла? Как она лечится?
Недостаточность миофосфорилазы (болезнь Мак-Ардла) характеризуется болезненными спазмами мышц, напряжением мышц при нагрузке, периодической миоглобинурией. Описан феномен «второго дыхания», когда симптомы исчезают после короткого отдыха и не возвращаются при возобновлении умеренной физической нагрузки. Отсутствие миофосфорилазы блокирует метаболизм углеводов, и для восполнения энергетических трат как при нагрувке, так и в покое используются липиды. Симптомы развиваются в силу того, что этот источник энергии при интенсивной нагрузке оказывается недостаточным.
Пациента прежде всего следует предупредить о риске рабдомиолиза, который может быть спровоцирован физической нагрузкой. Его также следует проинструктировать о важности изменения стиля жизни, о необходимости исключить интенсивные физические нагрузки, важности немедленного обращения к врачу при развитии миоглобинурии. Лечение направлено на обход биохимического блока путем снабжения мышцы промежуточными продуктами гликолиза (глюкоза, фруктоза), что приводит к увеличению работоспособности у некоторых пациентов, но их долговременное использование приводит к нежелательному увеличению веса и оказывается малоэффективным.
19-летнего юношу, ранее отмечавшего интенсивные болезненные мышечные спазмы при физической нагрузке, инструктор в военном лагере заставил пробежать 50 пролетов лестницы. Спустя несколько часов он отметил появление темной мочи (цвета кока-колы), лихорадку, озноб и сильную болезненность мышц. Анализ крови показал нормальное содержание лейкоцитов, умеренную анемию и увеличение количества ретикулоцитов. Проведите дифференциальный диагноз. Наиболее вероятно, что у пациента недостаточность фосфофруктокиназы (ФФК) (гликогеноз VIII типа, или болезнь Таруи). Он может страдать и болезнью Мак-Ардла, но гемолитическая анемия с увеличением числа ретикулоцитов более характерна для недостаточности ФФК. В норме эритроцитарная ФФК состоит из двух субъединиц: мышечного типа (М) и эритроцитарного типа (R). У пациентов с недостаточностью ФФК отсутствует субъединица М. Тип наследования в большинстве случаев—аутосомно-рецессивный. Снижение активности ФФК в эритроцитах может быть выявлено у лиц, не имеющих каких-либо симптомов. Недостаточность мышечной ФФК приводит к блокаде гликолиза. Клинические проявления недостаточности ФФК очень близки симптомам болезни Мак-Ардла и включают снижение толерантности к физической нагрузке, которая проявляется при интенсивных физических упражнениях быстрым мышечным утомлением, напряжением и болью в мышцах, которые сохраняются в течение нескольких минут или нескольких часов.
39. Каковы клинические признаки миопатии, связанной с недостаточностью карнитина?
Карнитин играет существенную роль в метаболизме жирных кислот в мышечном волокне. Недостаточность карнитина вызывает медленно прогрессирующую слабость мышц конечностей, шеи и туловища, начинающуюся в раннем детском или среднем возрасте. Кроме того, могут наблюдаться кардиомиопатия (иногда фатальная) и полиневропатия, но миалгии и мышечные спазмы редки. Уровень КФК нормальный или умеренно повышен. Уровень карнитина (свободного и ацилированного) обычно снижен в мышце, но не в сыворотке или печени. В мышечных волокнах отмечается большое количество липидных гранул. Регулярный прием L-карнитина (50-100 мг/кг/день внутрь в несколько приемов) приводит к увеличению мышечной силы. У некоторых пациентов может отмечаться улучшение при применении рибофлавина и преднизолона.
![]()
- Категория: Секреты неврологии
33. Существует ли связь между злокачественной гипертермией и болезнью центрального стержня?
Болезнь центрального стержня — это врожденная миопатия. Злокачественная гипертермия-это реакция на общую анестезию (наркоз). Оба состояния передаются по аутосомно-доминантному типу, а гены этих заболеваний располагаются друг за другом в хромосоме 19 (19ql2-ql3,2). В1993 году у пациентов со злокачественной гипертермией и болезнью центрального стержня были выявлены мутации рианодинового рецептора 1 типа, которые были выявлены как при болезни центрального стержня, так и при злокачественной гипертермии. Некоторые пациенты с болезнью центрального стержня подвержены злокачественной гипертермии. Они и члены их семей должны быть предупреждены о возможности развития злокачественной гипертермии как реакции на анестетики.
34. Какие симптомы должны наводить на мысль о метаболической миопатии?
Клинические проявления, подозрительные на метаболическую миопатию, включают мышечную боль при физической нагрузке, напряжение мышц или болезненные спазмы (крампи) и миоглобинурия. Некоторые заболевания связаны со стабильной или прогрессирующей мышечной слабостью.
35. Как классифицируют метаболические миопатии?
Метаболические миопатий выделяют в соответствии с тем, какой из путей метаболизма нарушен.
Гликоген метаболизируется до молочной кислоты или пирувата, обеспечивая мышцы энергией как при интенсивной нагрузке, так и в анаэробном состоянии. В аэробных условиях пируват включается в цикл трикарбоновых кислот, чтобы генерировать больше энергии путем окислительного метаболизма.
Метаболизм липидов служит источником энергии в покое и при длительной субмаксимальной нагрузке. Жирные кислоты с длинными цепями переносятся в митохондрии, где карнитинпальмитоил-трансфераза (КПТ) катализирует реакцию с образованием ацилкарнитиновых эфиров, которые окисляются в ацетил-Ко А и АТФ. Ацетил-КоА далее участвует в цикле трикарбоновых кислот.
Фосфокреатин и пурййовый нуклеотидный цикл используются во время коротких высокоинтенсивных нагрузок. Фосфокреатин восстанавливает уровень АТФ из АДФ, а КФК—это фермент, который катализирует перенос фосфатных групп в этих реакциях.
Митохондрии вырабатывают ферменты, которые влияют на окисление пирувата, глюкозы и жирных кислот в аэробных состояниях, формируя градиент ионов водорода.
36. Что такое недостаточность кислой мальтазы? Как проводить дифференциальный диагноз?
Недостаточность кислой мальтазы (гликогеноз II типа)—аутосомно-рецессивное заболевание, связанное с недостаточностью лизосомного фермента альфа-гли-козидазы (кислой мальтазы). Она может проявляться в любом возрасте и имеет три различные формы: инфантильную (болезнь Помпе), детскую и взрослую. Взрослая форма проявляется на третьем-четвертом десятилетиях жизни постепенно нарастающей безболезненной слабостью в тазовом и плечевом поясе, а также конечностях. Ее часто ошибочно принимают за ПМ, болезнь двигательных нейронов, миотони-ческую дистрофию или конечностно-поясную форму мышечной дистрофии.
Могут диспропорционально тяжело поражаться дыхательные мышцы. Уровень КФК обычно умеренно повышен (в 2-10 раз выше нормы). Остается в норме образование лактата—нормальное. ЭМГ показывает большое количество сложных повторяющихся разрядов в добавление к миопатическим изменениям и спонтанной активности. Если явные разряды отсутствуют, картина ЭМГ напоминает полимиозит. Таким образом, для постановки диагноза может потребоваться биопсия мышцы. Характерные признаки те же; что и при вакуолярной миопатий. Вакуоли содержат ШИК-положительный материал с выраженной активностью кислой фосфатазы. Похожие вакуоли встречаются при хлорохиновой миопатий. Эффективное лечение отсутствует, хотя диета с высоким содержанием белка и низким содержанием углеводов приводит к некоторому улучшению.
![]()
Новости медицины
Много миллионов человек в мире принимают статины, но исследования показывают, что только 55% из тех, кому рекомендуется принимать статины, принимают их. Это большая проблема, потому что исследования также показывают, что те из группы...
Связанное с беременностью высокое кровяное давление может привести к долгосрочным сердечным рискам, показывают новые исследования.
Кэролин Консия, столкнулась с более серьезными последствиями репрессий против назначения опиоидов, когда узнала, почему сын ее подруги покончил с собой в 2017 году.
Новое исследование показывает, что психические заболевания не являются фактором большинства массовых расстрелов или других видов массовых убийств.


{kind=link}